









“4+7”后避价格战!看扬子江等如何布局新3类药
“4+7”药品带量采购后,许多生产厂家的产品将要面临价格战。不少企业考虑和研究布局新3类。那么,目前新3类的注册和上市情况是怎样的呢?
2018年,首次有产品以化学药品新注册分类3类上市获批。2018年国家药品监管局共批准了化学药品新3类产品3个,且都是注射剂。除了布洛芬注射液之外,氟比洛芬酯注射液和盐酸右美托咪定注射液都已有国产生产厂家在国内上市。
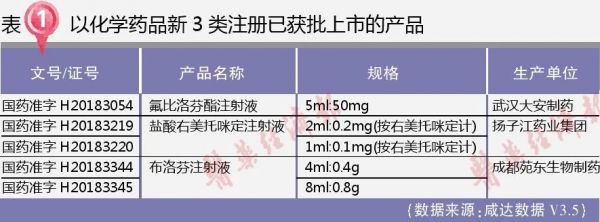
本文将回顾这3个产品的上市历程,了解目前已上市的新3类产品在注册历程中的临床研究状况,为未来的相关注册提供启示。
新3类政策细节三问
问1:原研药品在国外的申报资料是否完善?
根据化学药品新3类定义,新3类药品应与原研药品的质量和疗效一致。原研药品指境内外首个获准上市,且具有完整和充分的安全性、有效性数据作为上市依据的药品。

这意味着如果原研药品在国外的申报资料不完善,有可能在国内就要做验证性临床试验。
问2:是否需要做人体药代动力学研究和随机对照临床试验?
化学药品新注册分类新3类药品和新4类一样,申请人进行自我评估后,在能够确保受试者安全的前提下,可进行BE试验备案,待完成BE试验后,直接提出上市注册申请。
根据2007年版《药品注册管理办法》(局令第28号),旧3类并不需要做生物等效性试验,但旧3类药需进行人体药代动力学研究和至少100对随机对照临床试验。多个适应症的,每个主要适应症的病例数不少于60对。避孕药应当进行人体药代动力学研究和至少500例12个月经周期的开放试验。只有局部用药且仅发挥局部治疗作用的制剂、不吸收的口服制剂两种情况可免予进行人体药代动力学研究。
旧6类中的口服固体制剂才需要进行生物等效性试验,一般为18~24例,但是需要用工艺和标准控制药品质量的,应当进行临床试验,临床试验的病例数至少为100对。
因此,新3类完成生物等效性试验之后,是否还需要进行100对随机对照临床试验,是大家所关注的。
问3:是否需要补人种差异相关研究?
2018年10月,国家药监局发布《接受药品境外临床试验数据的技术指导原则》,对有效性和安全性数据有了定义。
有效性数据,主要包括境外关键临床试验数据和在中国开展的临床试验数据,既要从整体上确证研究药物的有效性,还要分析中国亚组与总体人群的一致性。
安全性数据,包括境内外所有用于安全性评价的数据,既要分析总体安全性,还要分析中国亚组与总体人群的一致性。
除了上述的境外临床试验数据的有效性和安全性评价,药品注册申请人还要符合中国药品注册管理要求,在汇总境内外各类临床试验的临床试验数据包进行完整分析的基础上,对关键的临床试验数据进行评价,以确证研究药物的有效性,并且遵循ICH关于接受国外临床资料的种族影响因素(E5)要求,分析中国亚组与总体人群的一致性,以支持境外临床试验结果外推至中国人群。这意味着如果进口药品没有人种差异的研究,新3类产品注册时需要补人种差异相关的研究。
获批产品上市经验
氟比洛芬酯注射液(武汉大安)
关键注册信息:
◎人体药代动力学研究(旧3类要求)
◎随机对照临床试验(旧3类要求)
◎生物等效性试验(旧6类要求)
根据《中国上市药品目录集》,2004年上市的北京泰德制药股份有限公司的氟比洛芬酯注射液可视为参比制剂和标准制剂。
武汉大安的氟比洛芬酯注射液2010年是以旧6类申报,2013年获批临床,2013年和2014年共启动了两个临床,分别是24例的人体生物等效性试验和240例的Ⅲ期随机、双盲、阳性药平行对照、多中心临床试验。
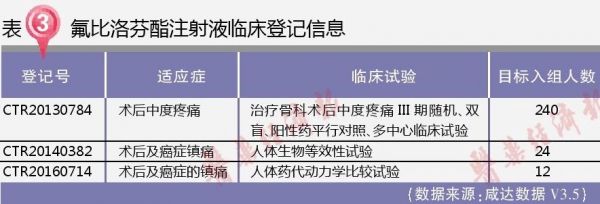
2014年以旧6类继续申报生产,后遇2015年临床自查核查,被迫公告撤回。
2016年补做人体药代动力学比较试验后,2017年武汉大安以新3类再次申报生产氟比洛芬酯注射液,2017年以“申请人主动撤回并改为按与原研药质量和疗效一致的标准完善后重新申报的仿制药注册申请”为由进入CDE第二十二批拟纳入优先审评名单。
2018年3月,武汉大安的仿制药获批,获批的适应症为术后及癌症的镇痛,与北京泰德获批的适应症一致。
2018年,氟比洛芬酯注射液补充申请,已经被临床试验默示许可,获批研究的适应症为术后及癌症的镇痛,但暂未查询到相关的临床研究,预计属于上市后再研究的临床观察。
综上所述,武汉大安的氟比洛芬注射液做了旧3类要求人体药代动力学研究和随机对照临床试验和旧6类的生物等效性试验才得以获批上市。
盐酸右美托咪定注射液(扬子江)
关键注册信息:
◎大量提供国外研究信息(包括不良反应、药物相互作用和药代动力学等)
扬子江可以查询到的信息是自2016年起就以新3类申报生产,暂未查到相关的临床研究信息。
2018年6月获批,适应症为用于行全身麻醉的手术患者气管插管和机械通气时的镇静;用于重病监护治疗期间开始插管和使用呼吸机病人的镇静,连续输注不可超过24小时。
从《中国上市药品目录集》公布对扬子江的盐酸右美托咪定注射液的说明书来看,扬子江的申报资料提供了很多国外研究报道的信息,包括不良反应、药物相互作用和药代动力学。
2016年扬子江还申报了盐酸右美托咪定氯化钠注射液上市,也是以新3类申报,2018年3月的审评结论是批准临床,但尚未查询到相关的临床研究信息。
若扬子江真的没有完成临床试验就获批上市,预计和扬子江大量提供了国外的临床综述,以及右美托咪定属于指南一线用药、临床疗效确切安全有关。
布洛芬注射液(成都苑东)
关键注册信息:
◎人体药代动力学研究(旧3类要求)
◎随机对照临床试验(旧3类要求)
◎生物等效性试验(旧6类要求)
布洛芬(Ibuprofen)是环氧化酶抑制剂,为非甾体类解热镇痛药(NSAIDs),具有解热、镇痛及抗炎作用。该品种由英国Boots公司研制,并于1969年首次在英国上市。上市后的几十年中,国内外主要使用的为口服和局部给药剂型,直至2009年6月11日,美国FDA批准Cumberland Pharmaceuticals公司研制的布洛芬注射液上市,该品种才作为静脉给药的注射剂型上市。
成都苑东和四川阳光润禾在2013年起以化学药品旧3.3类报产布洛芬注射液,4个受理号都在2015年临床自查核查公告撤回。从药物临床试验登记与信息公示平台可得,成都苑东的布洛芬注射液在2013年启动了人体药代动力学研究、随机对照临床试验和旧6类的生物等效性试验。

2016年成都苑东以新3类重新报产,2018年7月获批生产,获批适应症是用于成人治疗轻至中度疼痛,作为阿片类镇痛药的辅助用于治疗中至重度疼痛,以及用于成人发热的退热治疗。
2018年9月申请补充申请,目前补充申请在审评审批中状态。
此外,2014年成都苑东以新2类申报右旋布洛芬注射液,2016年6月已获批临床,但是暂未公开临床研究信息。
小结<<<
“4+7”药品带量采购后,许多生产厂家的产品将要面临价格战,不少企业考虑布局新3类。但是,对于新3类产品是否需要启动临床研究,尚未有定论。
从目前获批的产品看,除了盐酸右美托咪定注射液是国内外上市多年的产品尚未确定注册申报是否有启动临床外,其余2个获批的新3类产品都完成了人体药代动力学研究、随机对照临床试验和的生物等效性试验才获批上市。这意味着新三类的申报的第一家大概率都需要完成相关临床试验,但是第二家上市的企业是否也要完成相关研究仍待观察。
此外,2018年获批的产品都是注射剂,口服药的获批仍是空白,口服药的标准是否会和注射剂一致,仍待2019年有新产品获批后再分析。
责任编辑:露儿
 医院新规:查医药代表 挂钩产品
医院新规:查医药代表 挂钩产品大医院严查医械代表再升级!私下接触医务人员,直接停止采购公司代理产品。...
 编外人员被收保证金?医院取消编制大势所趋
编外人员被收保证金?医院取消编制大势所趋看到一家县级医院向编外人员收取5000元工作保证金,限时不交清者,医院不再使用,老徐认为:编制制度或早已不适应医院发展需要了。...
 三甲医院:8个药询价 要求至少稳定供货半年
三甲医院:8个药询价 要求至少稳定供货半年三甲医院:供货不稳定,踢出一年。...
 5省497名执业药师挂证被查实
5省497名执业药师挂证被查实今年“3•15”后,执业药师“挂证”问题引起全社会关注,按照国家药监局要求,自2019年5月1日起,各省级局组织对行政区域内的药品零售企业开展监督检查。...
 又一大药陷入致癌风波 多家外企全球召回产品!
又一大药陷入致癌风波 多家外企全球召回产品!一些雷尼替丁药物又被查实含有NDMA杂质,目前山德士、葛兰素史克、印度瑞迪博士药厂均已停止雷尼替丁的供应并召回!...
 大洗牌!国务院检查组,进入医疗器械企业了
大洗牌!国务院检查组,进入医疗器械企业了国家严惩在医疗器械购买、销售、纳税...等多个环节的违规情况。...
 4+7全国扩围,中标结果流出
4+7全国扩围,中标结果流出大跌眼镜!4+7全国扩围结果出炉,有外企低价入围,部分原中选药企落标,多个品种再次刷新底价!...
 国务院发文:医械行业,筛选重点企业监管
国务院发文:医械行业,筛选重点企业监管当监管方式越来越科学,不合规或打擦边球的械企面临的压力就越来越大。...
 报告显示:多数医生不再愿意接待医药代表
报告显示:多数医生不再愿意接待医药代表报告显示,医生不再愿意接待医药代表,医药代表的必要性在降低。...
 750家医药企业,最新离职率公布
750家医药企业,最新离职率公布2019年1月到6月的市场薪酬数据白名单公布,其中医药行业的行业增长和薪酬增长都维持稳定的高位;此外,报告采集了750家医药公司的数据,医药人的平均离职率仅为5.91%,为所有行业中最低的。...
 63个药,底价曝光(附名单)
63个药,底价曝光(附名单)(9月6日),山东省药品集中采购网发布《关于山东省药品集中采购拟备案采购产品最低外省及拟挂网价格公示的通知》(以下简称《通知》)。...
 医药业平均月薪公布 仍是最好的就业去向之一
医药业平均月薪公布 仍是最好的就业去向之一据东方财富Choice数据,医药行业平均月薪为1.03万,同比涨幅较大,为11.75%,仅次于公用事业等行业,排名第六名。...